Materials Innovations
Full Html
Vol 1 Issue 1
Axial Modified Derivatives of Subphthalocyanine for Small Molecule based Organic Solar Cells
Pages: 1-12
Doi:
Doi URL: http://doi.org/10.54738/MI.2021.1102
Iza Shahid 1 , Faiza Jilani 1 , Amna Ayub 1 , Saleem Iqbal 2 , Umer Yaqoob 1 , Javed Iqbal ?? 1
1 Department of chemistry, University of Agriculture, Faisalabad, 38000, Pakistan
2 Department of Chemical Engineering, Punjab Bioenergy Institute, Wah Engineering College, University of Wah, Wah Cantt, Pakistan
3 University of Agriculture, Faisalabad, 38000, Pakistan
Four new molecules namely BsubPcM1, BsubPcM2, BsubPcM3 and BsubPcM4, are designed by substituting different electron withdrawing groups as acceptor groups at boron atom (axial substitution) of subphthalocyanine i.e., cyanide (BsubPcM1), 1,2,3,4,5-pentafluoro-6-metoxybenzene (BsubPcM2), 2-methylisoindoline-1,3-dione (BsubPcM3) and 1-methyl-4-(prop-1-yn-yl) benzene (BsubPcM4). Photovoltaic parameters are estimated through DFT by employing selective method B3LYP. Frontier molecular orbitals, Density of states, Optoelectronic studies, and transition density matrix calculations are employed to characterize the chemical reactivity, charge mobility, open circuit voltage and maximum absorption of all designed molecules. The reorganization energy calculations proposed that the electron mobility for BsubPcM1 (0.0093), BsubPcM2 (0.0077), and BsubPcM4 (0.0095) is smaller than BsubPc-Cl (0.0097). Furthermore, BsubPcM1 and BsubPcM4 carry the least value of hole reorganization energy (0.0042) and (0.0039) respectively which is less than the Parent molecule BsubPc-Cl (0.0044) that symbolize the magnified hole transfer. Amongst all, BsubPcM3 has maximumr wavelength of absorption (577.9 nm) with minimum bandgap (2.46 eV). The results show that axial modification of Boron Subphthalocyanine Chloride molecule is a promising pathway for the development of modified efficient D-A type materials for photovoltaic applications.
Keywords
Subphthalocyanines, Optoelectronic properties, Charge mobility, Transition density matrix, Density of states
The field of organic photo electronics is drawing massive attention because of its potential to provide cost effective, simple and tunable structural properties for energy production. In an organic photovoltaic devices, the most prominent factor Jsc (short circuit current density) can be improved by controlling the morphology and structural properties of the material.1 The open circuit voltage mainly depends upon the type of material used in organic solar cell. Large number of commercially available materials have been reported which are modified as donor materials. But there is no such type of development for those materials that can act as an electron acceptors in a photovoltaic devises, except the fullerenes.2Although, fullerenes are genuine, efficient and glaring material for the transportation of electrons but this class of electron accepting material have major issues with long time stability and maximum obtained open circuit voltage bounded by narrow band gap.3 Although Fullerenes were used intensively due to their strong oxidization-reduction potential and structural properties but on the other side, phthalocyanines is a class of material that have an excellent light harvesting properties and dominant characteristics to behave like electron donor due to their strong redox chemistry. As the phthalocyanines generally maintain reorganization energies of fullerenes that’s why it is recognized as an ideal material for the generation of long lived radical ion pair.4
The conjugated π electrons in macrocycle of phthalocyanines molecules are responsible for their effective absorption spectra in visible portion of electromagnetic spectrum. Subphthalocyanines are aromatic and non-planner molecule, having bowl shaped geometry in which three N-fused rings of di-iminoisoindole surround the central boron atom.5 The fourteen π electrons conjugated shows enhanced optical properties that are major contributor to their extensive use in optics, dyes and organic photo electronic devices. These properties can be modified by suitable substitution and derivatization at the axial and peripheral substituent. Subphthalocyanine molecule is chiral and it owned the C1 and C3 symmetry.6 By utilizing this symmetric feature of molecule, the development of complex molecules can be enhanced.7
It is noted that peripheral replacement of ligand has more strong and dominant effect on the redox potential of Subphthalocyanines as compared to axial derivatization. Hence redox behavior and stability of Subphthalocyanines is inevitable because of its long term stability for radical cation/anion, effective charge transportation, and orientation and configuration of energy levels of frontier molecular orbitals8.
Herein, we designed four new donor-acceptor type molecules (BsubPcM1-BsubPcM4) by replacing axial substituent Cl of BsubPc-Cl molecule with four different acceptor moieties. All proposed units have three subunits of isoindole rings as a central core with distinct electron-withdrawing acceptor groups projected outward. The idea behind the modification is to tune the optoelectronic properties of Subphthalocyanines and to observe the effect of these properties on the PCE of organic solar cells. Different parameters like, reorganization energy and binding energy of exciton, frontier molecular orbital analysis, absorption profile, transition density matrix, dipole moment, and device performance were calculated to check the efficiency of all the investigated molecules as an active and efficient candidate for organic photovoltaics (OPV).
In the present quantum mechanical report, molecular frameworks were drawn using GaussView 6.0.16. All the computational simulations were theoretically assessed with the assistance of Gaussian 09 W programs. Density functional theory (DFT) was chosen for geometry optimization9, four different computational methods such as B3YLP10, CAMB3LYP11, MPW1PW9112, and WB97XD13 in conjunction with 6-31G (d,p) basis set were used to obtain the optimized ground state geometries of Parent molecule BsubPc-Cl. Then the λmax value of Parent molecule was calculated through above-mentioned DFT method and compared with reported experimental absorption maxima value. A good agreement with experimental absorption values were exhibited by B3LYP functional with 6-31G (d,p) basis set, so this functional was selected for further calculations.
Absorption spectra of all the optimized molecules were determined first in the gas phase, and then in solvent (toluene). The polarizable continuum model, using the integral equation formalism (IEFPCM) was applied to assess the solvent effect.14 By employing the same suggested method, density of states (DOS) calculations were executed, and the results were interpreted by the pyMOlyze program.15 Furthermore, reorganization energies of anion and cation, charge transfer analysis, open circuit voltage, frontier molecular orbital (FMO) analysis and transition density matrix (TDM) of all the investigated molecules were performed by using B3LYP method combined with 6-31G (d,p) basis set.
Generally, reorganizational energy is classified into two parts, internal reorganization energy (λint) and external reorganization energy (λext). The (λint) deals with the rapid rate of changes in geometry while the later explain the effects of the external environment. It is reported that the rate of charge transfers and λint, have a direct relationship.16 Here, in continuing research we neglected the external environmental effect and solely considered the internal effect.17 Therefore, for electrons and holes, the internal reorganization energies were calculated by using cited equations (1) and (2).18
1 λe= E0- -E- +E-0 -E0
2 λh= E0+ -E+ +E+0 -E0
Where, E-° , E+° refers to the energy of anion and cation, respectively, computed from anion and cation of molecule. E°- and E°+ reflect the energy of anion and cation, respectively, calculated from optimized neutral molecules. E? serves as ground state energy of neutral molecule. At the same time, both E+ and E- are cationic and anionic energies, respectively, calculated from geometrically optimized molecules of cation and anion.
In our research, Boron subphthalocyanine chloride molecule was taken as reference. The reference molecule contains particular macrocyclic ring, organized by three subunits of isoindole that are linked by three aza bridges, and a boron atom present in the central binding core. We replaced the axial substituent of reference molecule with four different acceptor groups, in order to magnify the photo physical properties of Parent molecule BsubPc-Cl. By doing modifications in parent molecule (BsubPc-Cl), we obtained four distinct molecules named BsubPcM1, BsubPcM2, BsubPcM3 and BsubPcM4 as shown in Figure 1. The configured geometries of all the proposed units at B3YLP/31G (d, p) are demonstrated in Figure 2.
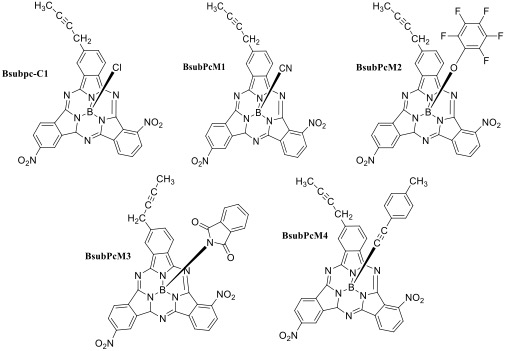
Figure 1: Molecular framework of parent and all proposed molecules.
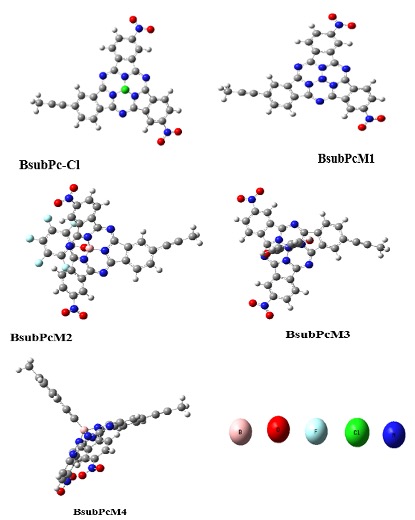
Figure 2: Optimized geometries of all investigated molecules at B3YLP/6-31G (d, p).
To check the electrical and optical characteristics of Parent molecule BsubPc-Cl, four computational methods B3LYP, CAMB3LYP, MPW1PW91 and, WBP7XD were employed along with 6-31G (d,p) basis set. The maximum absorption of BsubPc-Cl molecule calculated with four computational methods B3LYP, CAMB3LYP, MPW1PW91 and, WBP7XD were 570 nm, 526 nm, 547 nm, and 525.4 nm, respectively. According to literature, reported experimental value of BsubPc-Cl was 596 nm19. Graphical presentation of all results with stated methods are shown in Figre 3. After careful assessment, we observed that the functional B3LYP has a very close match to the experimental results. Hence, the functional B3LYP/6-31G (d, p) was selected to study the further properties of our systems.

Figure 3: Absorption profile of Parent moleculeBsubPc-Cl with the CAMB3LYP, MPW1PW91, B3LYP and WB97XD functional with 6-31G (d, p) basis set.
For the understanding of optoelectronic properties of organic solar cells (OSCs), frontier molecular orbital (FMO) diagrams are crucial. The charge transition in organic solar cells varies remarkably by the distribution pattern of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). The conduction band theory reveals the dynamic character of HOMO and LUMO by taking ground state (HOMO) for conduction band and excited state (LUMO) for valance band. The energy difference of these two orbitals is called energy bandgap (Eg). The energy bandgap offers a useful insight to study exciton dissociation, open circuit voltage, and provide a clear intimation about the performance of photoactive material. A material that has a low band gap with high power conversion efficiency is convenient for photovoltaic OSCs. So, we characterized the optical properties, conducting behavior and electron density of all designed molecules by using FMOs. The distribution patterns of HOMO and LUMO by the frontier molecular orbital diagrams (FMOs) are presented in Figure 4, and energies of HOMO & LOMO with energy bandgap of all the designed molecules and parent molecule are tabulated in Table 1.

Figure 4: FMOs of Parent molecule and all proposed units.
Table 1: Energy of HOMO, energy of LUMO and band gap of all molecules at B3LYP with same basis set.
|
Molecules |
E HOMO |
E LUMO |
E g |
|---|---|---|---|
|
BsubPc-Cl |
-5.96 |
-3.47 |
2.49 |
|
BsubPcM1 |
-6.04 |
-3.56 |
2.48 |
|
BsubPcM2 |
-5.95 |
-3.46 |
2.49 |
|
BsubPcM3 |
-5.73 |
-3.26 |
2.46 |
|
BsubPcM4 |
-5.75 |
-3.28 |
2.47 |
hows the value of HOMO (-5.95 eV) and LUMO (-3.46 eV) along with energy bandgap (2.49 eV) for parent molecule BsubPc -Cl. For all other designed molecules (BsubPcM1-BsubPcM4), HOMO values were found to be -6.0349 eV, -5.9489 eV, -5.7277 eV and -5.7484 eV and the LUMO were located at -3.5589, -3.4563, -3.2634 and, -3.2751(eV) respectively.
Energy difference (H-L) is a significant element for efficient charge transfer. A small energy difference leads to high charge transfer. BsubPcM1-BsubPcM4 exhibit H-L energy difference of 2.47, 2.492, 2.46, and 2.47 (eV), respectively. Among all designed molecules, BsubPcM3 have shown narrow band gap, and this might be due to 2-methylisoindoline-1,3-dione acceptor group, which provides the extended conjugation in the molecule. On the other hand, the energy bandgap for BsubPcM1 and BsubPcM4 is much appreciable than BsubPc-Cl due to the presence of electron-withdrawing group cyano-benzene and toluene in acceptor part of the molecule, which cause extended conjugations in whole molecule. The descending order of HOMO & LUMO energies of all the investigated molecules, including parent and designed molecules is BsubPcM3>BsubPcM4> BsubPcM2>BsubPc-Cl> BsubPcM1 and the descending order of bandgap energy of all molecules is BsubPcM3> BsubPcM4> BsubPcM1> BsubPcM2= BsubPc -Cl. Hence, from the above discussion it is concluded that BsubPcM3 as an efficient candidate in term of optoelectronic properties due to its narrow bandgap for OSCs.
Density of states (DOS) help to explain the particulars, propound by FMOs. So, to support FMOs and further analysis for electronic properties of all the molecules, the density of state (DOS) analysis in association with frontier molecular orbital was carried out at B3LYP/6-31G(d, p) (Figure 5). Like a single sheet, the donor part of designed molecules (BsubPcM1-BsubPcM4) arranged in one plane which promotes the flow of charge, and the acceptor part is projected outward. The results of DOS analysis described the switching of electron density around HOMO-LUMO by the acceptor moieties with different electron withdrawing groups. For reference molecule BsubPc-Cl and designed molecule BsubPcM1, the electron density is distributed mainly around the central binding core of the molecule. Similarly, LUMO resides mainly on the central core and a bit on isoindole rings and side groups. The distribution model of BsubPcM2 is somewhat different, In BsubPcM2 HOMO is more focused on the electron-rich central core, isoindole rings, and a small density located on the acceptor part. For BsubPcM3 and BsubPcM4 the distribution pattern is the same. In both molecules, HOMO is localized strongly on central binding core, and LUMO is localized on the subunits of isoindole rings, and a bit of it resides on side groups. From DOS analysis it is evident that for all proposed molecules (BsubPcM1-BsubPcM4) and reference molecule BsubPc-Cl, electron density is significantly delocalized and (HOMO-LUMO) distribution scheme is analogous to the distribution pattern determined by B3LYP/6-31G (d, p) method, as shown in Figure 4.

Figure 5: DOS (density of states) of parent molecule BsubPc-Cl around HOMO and LUMO and designed molecules BsubPcM1-BsubPcM4 at selected computational method.
Organic chromophores mostly show absorption in UV/visible region of the spectrum, and their absorption profiles helps to evaluate their optical properties. Optical properties of all the investigated molecules were analyzed by using B3YLYP/6-31G (d, p) level of theory, in an organic solvent (toluene). Experimentally recorded value of parent molecule BsubPc -Cl and computed λmax values of all the designed molecules along with dipole moment, oscillator strength and concerned assignments are listed in Table 2.
Table 2: Absorption parameters of all the proposed molecules at B3LYP/6-31G (d, p) method.
|
Molecule |
Calc. λ max (nm) |
Oscillator strength |
Assignments |
Dipole moment (D) |
|---|---|---|---|---|
|
BsubPc-Cl |
570.5 |
0.4576 |
H→L (97%) |
7.32 |
|
BsubPcM1 |
574.5 |
0.4417 |
H→L (97%) |
7.84 |
|
BsubPcM2 |
570.2 |
0.4418 |
H→L (97%) |
7.91 |
|
BsubPcM3 |
577.9 |
0.4432 |
H→L (97%) |
7.86 |
|
BsubPcM4 |
577.2 |
0.4463 |
H→L (97%) |
7.51 |
At the TDSCF/toluene/B3LYP/6-31G (d, p), all molecules, including the parent molecule show maximum absorption in the region of 300nm to 600nm. The simulated absorption spectra of all the investigated molecules are shown in Figure 6. From calculated data, the absorption profile of BsubPcM2 agrees with BsubPc-Cl with deviation of 0.3nm. BsubPcM3 tends to show strong absorption at 577.9 nm due to effective electron withdrawing 2-methylisoindoline-1, 3-dione acceptor group and efficient intramolecular charge transfer. The tabular data exhibit that BsubPcM1, BsubPcM3 and BsubPcM4 have bathochromic shift as compared to parent molecule, with the deviation of 4, 7.4 and 6.7 (nm) respectively. Molecule BsubPcM2 have shown a slightly weak hypsochromic shift with the deviation of 0.3nm than the BsubPc-Cl molecule which is mainly attributed to polyfluorinated benzene (1, 2, 3, 4, 5-pentafluoro-6-metoxybenzene) acceptor group. Decreasing order of λ-max values for the reference molecule and all designed molecules is BsubPcM3 > BsubPcM4 > BsubPcM1 > BsubPc-Cl > BsubPcM2.

Figure 6: Absorption spectrum of all proposed units BsubPcM1-BsubPcM4 and parent molecule BsubPc-Cl in solvent (toluene) phase at B3LYP with basis set 6-31G (d, p).
While designing the organic solar cell, charge mobility is one of the significant parameters to assess the performance and working of efficient photovoltaic material. Charge mobility can be calculated effectively by reorganization energies of electron and hole. The reorganization energy holds an inverse relationship with charge mobility so, if a molecule has small reorganization energy values it will have higher-charge mobility. Reorganization energy values depend upon many factors, but it mainly depends upon the geometry of anion (electron mobility) and cation (hole mobility)20.
By equation (1) and (2), reorganization energy has two parts, internal reorganization energy (related to internal changes λint) and external reorganization energy (related to external changes λext). Here, in our study, we ignored the environmental relaxation and external changes as it does not contribute much and specifically, thus dealing with internal reorganization energy for our system. λint is the energy expense for geometry transformation from neutral to charged molecule and charged to neutral molecule.
The B3LYP computational method in conjunction with 6-31G (d, p) basis set employed to calculate the reorganization energies for our representative BsubPc -Cl and all designed molecules and the results are shown in Table 3. The value of λe (electron mobility) for parent molecule BsubPc-Cl is 0.0097113 eV, and theoretically computed values of λe for BsubPcM1-BsubPcM4 are 0.0093244, 0.0077906, 0.0101066 and 0.0095415 respectively. When designed molecules were compared with BsubPc-Cl, a representative molecule, it is found that BsubPcM1 (0.0093244), BsubPcM2 (0.0077906) and BsubPcM4 (0.0095415) has lowest value of λe. It means that these molecules have great ability of electron transfer between donor and acceptor parts. So, all investigated molecules can be arranged in following decreasing order of λe BsubPcM3 > BsubPc-Cl >BsubPcM4 > BsubPcM1 > BsubPcM2.
Table 3: Reorganization energies of all the molecules.
|
Specie |
λ e |
λ h |
|---|---|---|
|
BsubPc-Cl |
0.0097113 |
0.0044968 |
|
BsubPcM1 |
0.0093244 |
0.004205 |
|
BsubPcM2 |
0.0077906 |
0.0049494 |
|
BsubPcM3 |
0.0101066 |
0.0054715 |
|
BsubPcM4 |
0.0095415 |
0.0039971 |
The λh (hole mobility) of reference molecule is 0.0044968, and theoretically computed λh values for BsubPcM1-BsubPcM4 are 0.004205, 0.0049494, 0.0054715 and 0. 0039971, respectively Amongst all, BsubPcM4 has the least value of λh which express the best suitability of molecule as hole transporter between donor and acceptor part of the molecule. The descending order of hole mobility is BsubPcM3 > BsubPcM2 > BsubPc -Cl > BsubPcM1 > BsubPcM4. Approximately, all molecule showed low value of λh relative to BsubPc -Cl except BsubPcM3 and BsubPcM2 for them reverse is true. From the above interpretation, it is evident that BsubPcM2 dominant nominee for electron transfer and BsubPcM4 is best for hole transfer.
The transition density matrix is a useful approach to describe (a) The interaction of donor and acceptor groups (b) to predict the localization of electron-hole pair(c) to evaluate the electronic excitation. TDM provides characteristic and consistency length of any electronic transition between two states of the molecule and also regarded as best suited tool to explain the charge transfer excitations in photovoltaic materials. The computational hybrid functional B3LYP with6-31G (d,p) basis set was employed to examine the emission and absorption of all the investigated units in S1 (first excited state). As the hydrogen atom has a minimal contribution in transition, therefore they have been neglected by default.
For the evaluation of TDMs, all proposed molecules were divided into three parts, A (central core), B (Aromatic rings), and C (side groups). From TDM diagrams shown in Figure 7, it is evident that electron coherence (of all proposed unit and parent molecule) is the diagonal of part B (aromatic rings) and part A (central core unit). Electron coherence of C (side groups) is smaller as compared to other parts. Compared to parent molecule BsubPc-Cl, all the designed molecule showed high charge dissociation and current density (Jsc). The power conversion efficiency (PCE) and rate of charge dissociation of photoactive material is determined by binding energy of electron-hole pair (Eb)

Figure 7: TDM images of parent and proposed molecules BsubPcM1 to BsubPcM4 at the S1 state.
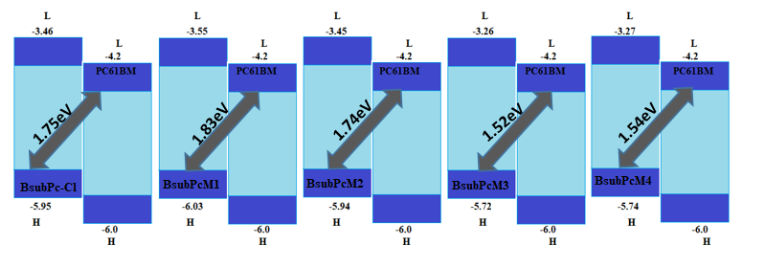
Figure 8: Energy bandgap values for all the investigated molecules with respect to acceptor polymer PC61BM.
3 Eb= Eg -Ex
Above equation was used to calculate the theoretical binding energies for all the investigated molecules in toluene solvent and results are listed in table 4.
Table 4: Binding, excitation and bandgap energies of Parent and proposed molecules.
|
Molecules |
E b |
E x |
E g |
|---|---|---|---|
|
BsubPc-Cl |
0.32 |
2.1731 |
2.49 |
|
BsubPcM1 |
0.33 |
2.1580 |
2.48 |
|
BsubPcM2 |
0.32 |
2.1742 |
2.49 |
|
BsubPcM3 |
0.32 |
2.1453 |
2.46 |
|
BsubPcM4 |
0.33 |
2.1482 |
2.47 |
In equation (3), Eg is energy band gap taken by EH-L and Ex is the difference of excitation energy from ground state to 1st excited state. Lower electron/hole interaction leads to lower value. To allow the dissociation of exciton into separate charges (hole and electron), exciton binding energy should be as low as possible. The binding energy of electron-hole pair of all the investigated molecules are comparable to each other’s and have following order BsubPcM1 = BsubPcM4 > R = BsubPcM2 = BsubPcM3 . This sequence indicates that among all molecules, BsubPcM3 and BsubPcM2 molecule have high rate of charge separation as they have low exciton binding energy. So, these molecules can show more significant, accessible, and smooth dissociation of exciton in excited state.

Figure 9: Charge transfer analysis betweenBsubPcM2 and PC61BM complex at B3LYP/6-31G (d,p).

Figure 10: Frontier molecular distribution schemes for molecule BsubPcM2 and polymer PC61BM.
The open circuit voltage (VOC) plays a vital role, as it can be used to estimate the operating mechanism and overall performance of the organic solar cell. Open circuit voltage is the overall amount of current that a PV device can produce at zero or null voltage. Voc is the measurement of the amount of recombination in the device, mainly it depends upon the saturation current and photo-generated current. In a molecule, the use of electron donor and acceptor groups with low energy bandgap values increase the absorption coefficient, which enhance the open circuit voltage and other light harvesting properties. A lower value of donor HOMO and higher value of acceptor LUMO leads to an efficient and effective open circuit voltage. In ongoing research, we worked on axially modified derivatives of Subphthalocyanines which is an electron donor, so we took the HOMO of our designed molecule and analyzed it with the LOMO of a famous and mostly used acceptor PC61BM21. Following equation was used to compute the open circuit voltage of all the investigated molecules.
Voc=EHOMOD - ELUMOA
By above equation computed VOC values were 1.75, 1.83, 1.74, 1.52, and 1.54 eV for BsubPc-Cl, BsubPcM1, BsubPcM2, BsubPcM3, and BsubPcM4, respectively (Figure 8). Among all newly designed molecules, BsubPcM1 exhibits high open-circuit potential relative to all other molecules. Hence, the fusion of our designed units with PC61BM acceptor will prove beneficial in the construction high PCE based solar cells.
In order to perceive the switching of charge density between best molecule among designed units and PC61BM, Charge transfer analysis was performed for instance. BsubPcM2 was selected as a donor with PC 61 BM (an acceptor polymer), as it has shown best photovoltaic and electrochemical properties i.e., low band gap energy (Eg), good absorption coefficient (λabs), best dipole moment(μ)and least value electron mobility(λe). The geometry optimization of BsubPcM2-PC 61 BM complex were performed at B3LYP/6-31G (d, p) level of theory. The optimized structure and orientation of the complex is shown in Figure 9. The optimized geometry of the BsubPcM2-PC 61 BM complex signified that, in terms of charge transfer, the orientation of BsubPcM2 and PC 61 BM is impressive. The molecule BsubPcM2 oriented towards PC 61 BM in a parallel manner. The dipole moment of the complex was calculated, the dipole moment’s direction has been suggested as an explanation for the successful and effective dissociation of exciton at the interface of complex BsubPcM2-PC 61 BM22 . The distribution pattern of frontier molecular orbitals for complex BsubPcM2-PC 61 BM was determined at same selected method and it is found that HOMO charge density located on the donor part (BsubPcM2) of complex while LUMO density resides on the acceptor (PC 61 BM) as shown in Figure 10. The description of orbital diagrams has shown that the shifting of electron density from electron donating to electron accepting part of complex is substantial evidence to consider our investigated molecules as an efficient candidate for the fabrication of OSCs.
Another powerful and scaling tool to scale the performance of OSCs is dipole moment. It has a compelling effect in the modeling of photovoltaic cells as it explains the solubility of photoactive material in solvent. A higher value of dipole moment leads to high solubility of material in an organic solvent like toluene. Therefore, efforts were made to calculate the dipole moment of designed molecules in solvent at same selected method. The values of dipole moment in toluene for all molecules were BsubPc -Cl (7.32) BsubPcM1 (7.84) BsubPcM2 (7.91) BsubPcM3 (7.86) and BsubPcM4 (7.51) as shown in Table 2. From table data it is very clear that all the engineered molecules have greater dipole moment than parent molecule, which means that all the designed molecules are capable of being soluble in any organic solvent. Molecule BsubPcM2 exhibits greatest dipole moment, attributed to the strong polarity effect of a highly electron-capturing (1, 2, 3, 4, 5,-Penta fluoro-6-methoxybenzene) acceptor group. The higher dipole moment amplified the self-assembly of compounds and makes a long chain, giving a stable path to charge transfer (if designed units have ability of packing). The overall order of dipole moment of proposed molecules, and reference molecule is following BusbPcM2 > BsubPcM3 > BsubPcM1 > BsubPcM4 > BsubPc-Cl.
In current studies, four D-A type small molecules were developed and reviewed theoretically for their successful and economical use in organic solar cells. The designed molecules studied by four different computational methods B3LYP, CAM-B3LYP, MPWIPW91, and WB97XD, with the basis set 6-31G (d, p). The electronic and optical properties of the proposed molecules were analyzed; these were found much better on the subject of their electronic structure and bandgap than the parent molecule (BsubPc-Cl). The maximum absorption (577.9nm) was observed by BsubPcM3, along with the lowest Eg (2.46 eV), credit to the 2-methylisoindoline-1,3-dione group. The hole mobility for BsubPcM4 and BsubPcM1 is smaller than BsubPc-Cl. The reorganization energy value for electron was found as BsubPc-Cl > BsubPcM4 > BsubPcM1 > BsubPcM2. The lower value of electron mobility concludes that designed molecules are best for electron transport material. Shifting of electron density by acceptor groups was studied by Density of states (DOS), which verified the shreds of evidence obtained from (HOMO & LUMO) quantum mechanical studies. The charge transfer analysis give a high value of Voc for BsubPcM1 with PC61BM indicates the successful moving of electron from donor to acceptor part of the complex. Hence, proposed molecules could be implemented to design efficient organic solar cells with broad absorption ranges, as they have excellent optoelectronic properties.
The authors acknowledge the financial and technical support from Punjab Bio-energy institute (PBI), University of Agriculture, Faisalabad (UAF), Pakistan.
Gier, H D De, Jahani, F, Broer, R, Hummelen, J C & Havenith, R W . 2016. Promising strategy to improve charge separation in organic photovoltaics: installing permanent dipoles in PCBM analogues. The Journal of Physical Chemistry A 120(27):4664–4671.
Cossi, M, Barone, V, Mennucci, B & Tomasi, J . 1998. Ab initio study of ionic solutions by a polarizable continuum dielectric model. Chemical Physics Letters 286(3-4):253–260.
Suboti?, B & Broni?, J . 2003. Theoretical and practical aspects of zeolite crystal growth. (pp. 174-278) CRC Press
Dennington, R, Keith, T A & Millam, J M . 2008. GaussView 5.0.8. In: GaussView 5.0.8. Wallingford: Gaussian. Inc
Sullivan, P, Duraud, A, Hancox, , Beaumont, N, Mirri, G, Tucker, J H, Hatton, R A, Shipman, M & Jones, T S . 2011. Halogenated boron subphthalocyanines as light harvesting electron acceptors in organic photovoltaics. Advanced Energy Materials 1(3):352–355.
Chai, J.-D & Head-Gordon, M . 2008. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Physical Chemistry Chemical Physics 10(44):6615–6620.
Claessens, C G & Torres, T . 2002. Synthesis, separation, and characterization of the topoisomers of fused bicyclic subphthalocyanine dimers. Angewandte Chemie International Edition 41(14):2561–2565.
Sullivan, P, Jones, T, Ferguson, A & Heutz, S . 2007. Structural templating as a route to improved photovoltaic performance in copper phthalocyanine/fullerene (C 60) heterojunctions. Applied Physics Letters91(23):233114.
Civalleri, B, Zicovich-Wilson, C M, Valenzano, L & Ugliengo, P . 2008. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 10(4):405–410.
Adamo, C & Barone, V . 1998. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The m PW and m PW1PW models. The Journal of chemical physics 108(2):664–675.
Sampson, K L, Jiang, X, Bukuroshi, E, Dovijarski, A, Raboui, H, Bender, T P & Kadish, K M . 2018. A Comprehensive Scope of Peripheral and Axial Substituent Effect on the Spectroelectrochemistry of Boron Subphthalocyanines. The Journal of Physical Chemistry A 122(18):4414–4424.
Yanai, T, Tew, D P & Handy, N C . 2004. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chemical physics letters. Chemical Physics Letters 393(1):51–57.
González-Rodríguez, D, Torres, T, Guldi, D M, Rivera, J, Herranz, M Á & Echegoyen, L . 2004. Subphthalocyanines: Tuneable molecular scaffolds for intramolecular electron and energy transfer processes. Journal of the American Chemical Society 126(20):6301–6313.
Khlyabich, P P, Burkhart, B, Rudenko, A E & Thompson, B C . 2013. Optimization and simplification of polymer-fullerene solar cells through polymer and active layer design. Polymer 54(20):5267–5298.
O'boyle, N M, Tenderholt, A L & Langner, K M . 2008. Cclib: a library for package-independent computational chemistry algorithms. Journal of computational chemistry 29(5):839–845.
Wei, G, Lunt, R R, Sun, K, Wang, S, Thompson, M E & Forrest, S R . 2010. Efficient, ordered bulk heterojunction nanocrystalline solar cells by annealing of ultrathin squaraine thin films. Nano letters10(9):3555–3559.
Burkett, S L & Davis, M E . 1995. Mechanisms of structure direction in the synthesis of pure-silica zeolites. 1. Synthesis of TPA/Si-ZSM-5. Chemistry of materials 7:920–928.
Ans, M, Iqbal, J, Eliasson, B & Ayub, K . 2019. Opto-electronic properties of non-fullerene fused-undecacyclic electron acceptors for organic solar cells. Computational Materials Science 159:150–159.
Jin, R & Chang, Y . 2015. A theoretical study on photophysical properties of triphenylamine-cored molecules with naphthalimide arms and different π-conjugated bridges as organic solar cell materials. Physical Chemistry Chemical Physics 17(3):2094–2103.
Köse, M E, Mitchell, W J, Kopidakis, N, Chang, C H, Shaheen, S E, Kim, K & Rumbles, G . 2007. Theoretical studies on conjugated phenyl-cored thiophene dendrimers for photovoltaic applications. Journal of the American Chemical Society 129(46):14257–14270.
González-Rodríguez, D, Claessens, C G, Torres, T, Liu, S, Echegoyen, L, Vila, N & Nonell, S . 2005. Tuning Photoinduced Energy-and Electron-Transfer Events in Subphthalocyanine-Phthalocyanine Dyads. Chemistry-A European Journal 11(13):3881–3893.
Ko?se, M E, Long, H, Kim, K, Graf, P & Ginley, D . 2010. Charge transport simulations in conjugated dendrimers. The Journal of Physical Chemistry A (12) 4388–4393.
Keywords: Subphthalocyanines, Optoelectronic properties, Charge mobility, Transition density matrix, Density of states
Materials Innovations (MI) is an interdisciplinary journal devoted to significant experimental and theoretical findings on the synthesis, structure, charachterization, processing and applications of materials. Materials Innovations is dedicated to publishing peer reviewed novel, cutting edge reports of broad interest to the materials science community.